Understanding Sickle Cell Anemia: Unraveling the Mysteries of a Genetic Disorder
Sickle cell anemia stands as a testament to the intricate relationship between genetics, biology, and health. This hereditary disorder, predominantly affecting individuals of African descent, has long fascinated and challenged scientists, clinicians, and affected individuals alike. From its discovery in the early 20th century to ongoing breakthroughs in treatment and management, sickle cell anemia remains a pivotal subject in both medical research and public health discourse.
The Genetic Basis
At the heart of sickle cell anemia lies a genetic mutation with profound physiological consequences. The disorder stems from a point mutation in the gene encoding the beta-globin subunit of hemoglobin, the protein responsible for transporting oxygen throughout the body. This single nucleotide change results in the substitution of a single amino acid in the hemoglobin molecule: glutamic acid is replaced by valine. This seemingly minor alteration triggers a cascade of events that ultimately leads to the characteristic sickle-shaped red blood cells.

The Sickle Cell Trait and Disease
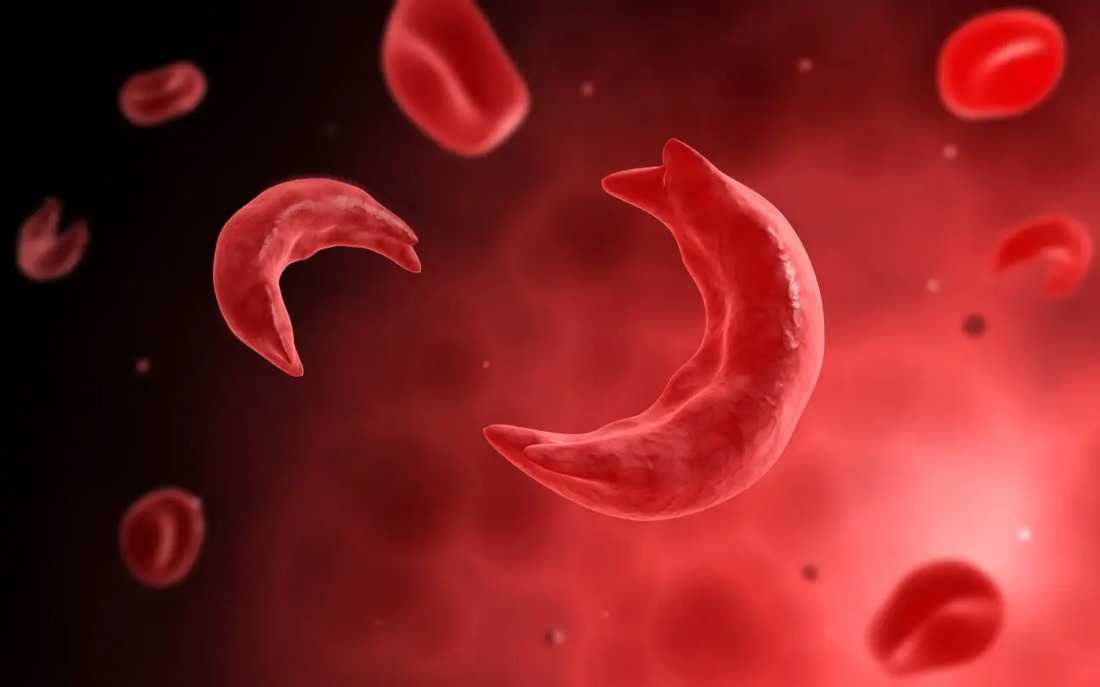
Individuals who inherit one copy of the mutated gene and one normal copy are said to have the sickle cell trait. While carriers of the trait typically do not display symptoms of the disease, they can pass the mutated gene to their offspring. On the other hand, individuals who inherit two copies of the mutated gene, one from each parent, develop sickle cell anemia. This double dose of the mutated gene leads to the production of abnormal hemoglobin, causing red blood cells to assume a rigid, crescent shape under certain conditions.
Clinical Manifestations
The hallmark of sickle cell anemia is its wide array of clinical manifestations, ranging from mild to severe. These include chronic anemia, vaso-occlusive crises, organ damage, and increased susceptibility to infections. The sickle-shaped red blood cells are prone to clumping together, obstructing blood flow and causing tissue ischemia and pain. Moreover, the compromised oxygen-carrying capacity of the abnormal hemoglobin predisposes individuals to tissue hypoxia, exacerbating the clinical burden of the disease.
Management and Treatment
Despite its complexity, sickle cell anemia is not without recourse. Over the years, advances in medical science have yielded various strategies for managing the disorder and improving patients’ quality of life. This includes supportive measures such as blood transfusions to alleviate anemia and hydration to prevent vaso-occlusive crises. Additionally, pharmacological interventions like hydroxyurea and newer agents targeting specific pathways in the disease process have shown promise in reducing symptom severity and frequency of complications.
Challenges and Future Directions
While considerable progress has been made in understanding and treating sickle cell anemia, significant challenges persist. Access to comprehensive care remains uneven, with disparities in healthcare infrastructure and resources exacerbating the burden of disease for marginalized populations. Moreover, the quest for a definitive cure continues, with ongoing research focusing on gene therapy, stem cell transplantation, and other innovative modalities.
Conclusion
Sickle cell anemia serves as a poignant reminder of the intricate interplay between genetics, biology, and health. From its humble origins as a curious hematological anomaly to its current status as a major public health concern, the disorder continues to captivate the scientific community and inspire hope for a brighter future. As we strive to unravel its mysteries and alleviate its burden, let us remain steadfast in our commitment to advancing the frontiers of medical knowledge and ensuring equitable access to care for all affected individuals.


