Sickle Cell Anemia: Understanding the Genetic Battle Within
Introduction
Sickle Cell Anemia is a complex and often misunderstood genetic disorder that affects millions of people worldwide. It is a condition characterized by misshapen red blood cells, causing a wide range of health problems. In this article, we will explore the basics of sickle cell anemia, its genetic basis, symptoms, treatments, and the ongoing research to improve the lives of those affected.
Understanding Sickle Cell Anemia
1. Genetic Basis:
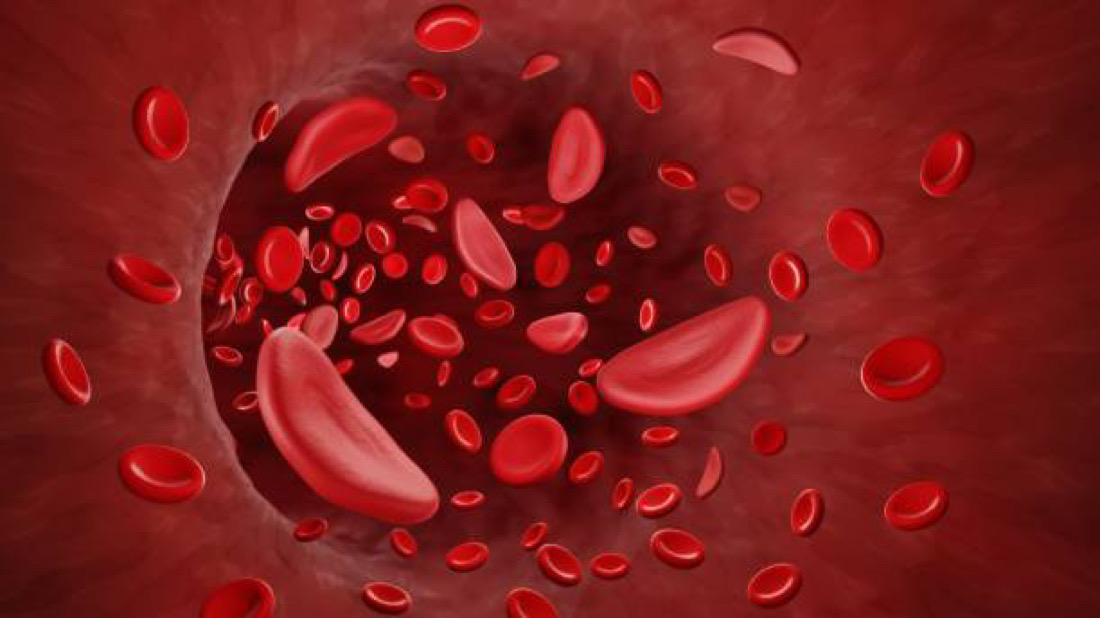
Sickle Cell Anemia is an inherited genetic disorder that primarily affects people of African, Mediterranean, Middle Eastern, and Indian descent. The condition is caused by a mutation in the HBB gene, which encodes a protein called hemoglobin. Hemoglobin is responsible for carrying oxygen from the lungs to the rest of the body.
In normal individuals, hemoglobin molecules are round and flexible, allowing red blood cells to move smoothly through blood vessels. However, in individuals with sickle cell anemia, a specific mutation leads to the production of abnormal hemoglobin known as hemoglobin S (HbS). When oxygen levels in the blood decrease, HbS molecules can stick together and form rigid, crescent-shaped cells, hence the name “sickle” cell anemia.
2. Symptoms:
The hallmark symptom of sickle cell anemia is pain. These pain episodes, known as “crises,” can vary in intensity and duration, and they occur when sickled red blood cells block blood flow in small vessels. Other common symptoms and complications include:
– Fatigue
– Anemia (a shortage of red blood cells)
– Jaundice (yellowing of the skin and eyes)
– Frequent infections
– Delayed growth in children
– Vision problems
– Organ damage (e.g., damage to the spleen, liver, and kidneys)
– Stroke
– Acute chest syndrome (a life-threatening complication)
3. Diagnosis and Treatment:
Diagnosing sickle cell anemia typically involves a blood test that checks for the presence of abnormal hemoglobin. Prenatal testing is also available for parents who carry the genetic trait to assess the risk of having a child with the condition.
Treatment options for sickle cell anemia aim to manage symptoms, prevent complications, and improve quality of life. These may include:
– Pain management with medications
– Blood transfusions to alleviate anemia
– Hydroxyurea, a medication that can reduce the frequency of pain crises
– Folic acid supplements to help produce healthy red blood cells
– Bone marrow or stem cell transplants (curative but risky)
Ongoing Research and Hope for the Future
Advancements in medical research and treatment options have improved the prognosis for individuals with sickle cell anemia. Recent developments include gene therapy and gene editing techniques that aim to correct the HBB gene mutation, potentially providing a cure for the condition.
Additionally, ongoing clinical trials and studies are exploring new therapies, including drugs that prevent sickling of red blood cells, improved pain management strategies, and innovative approaches to reduce the risk of complications.
Conclusion
Sickle Cell Anemia is a challenging genetic disorder that affects millions of people worldwide. While it can cause significant pain and health complications, advances in medical research and treatment offer hope for improved outcomes and, ultimately, a cure. Increased awareness, research funding, and support for individuals living with sickle cell anemia are essential steps toward a future where this condition no longer poses a significant threat to health and well-being.


